A major challenge of the sunflower genome project has been dealing with the large and repetitive nature of the genome. Below is a description of our custom annotation procedures and the results. You may skip to the results or download sections using the menu to the left.
Annotation procedures
The annotation procedures listed below were developed at UBC and INRA. The methodology and results on this page are for the HA412-HO genome.
Transposon Annotation
Given the complexity of the sunflower genome, the task of repeat annotation has been a major challenge in terms of not only the complex biology but also the computational time involved, which has required the a great deal of engineering to develop a system that scales with multi-gigabyte genomes. We put a significant effort towards developing a custom transposable element (TE) annotation approach that is both accurate and efficient, and this is implemented in the program Tephra which is now open source. This approach brings together many published tools and best practices to generate a set of TEs based on structural predictions and protein model support. We have also implemented programs to interfer patterns of demography and age, diversity, and DNA removal. A publication of this software is forthcoming so we will link to a broader discussion of the methods when that is published. For now, please refer to the project website for the software or the Downloads section for the transposon annotations.
Gene Annotation
The first step in our gene annotation process is to generate a masked genome with the transposons generated from the above steps using RepeatMasker with default settings. Gene models were produced by training several ab initio model-based gene prediction programs and incorporating these models, along with additional evidence, with MAKER-P to produce a single set of models.Initial steps - gene predictions
The first set of predictions were generated by the program EuGene and this work has been driven by collaborator Jerome Gouzy’s group at INRA Toulouse. EuGene integrates a wide range of evidence types (protein similarity and evidence of transcription), as well as different sources of predictions such as alternative gene splicing site predictors, start predictors, gene predictors and so forth. The sunflower EuGene pathway is shown in Figure 1.
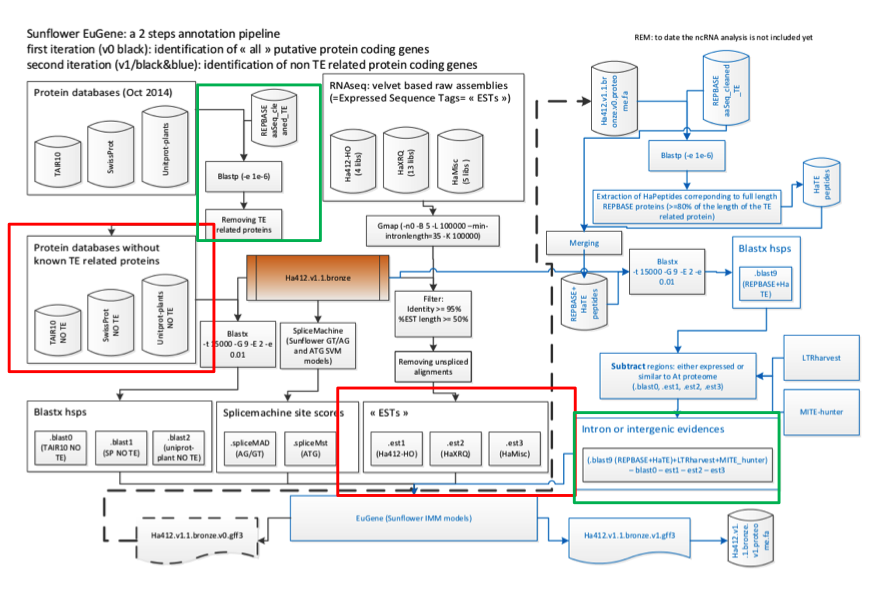
Figure 1. Sunflower Eugene Pipeline (courtesy of Sebastien Carrere)
Results from the annotation of the bronze assembly indicate that the gene space of sunflower is well-covered: 94.33 % of HA412-HO ESTs are correctly mapped and 90,935 protein coding genes are predicted, excluding transposable elements. Of these, 59,817 are supported by Full Length Best Hits (spanning 60% of the length of theA. thaliana | SwissProt | Unitprot_plant protein) or have EST/RNAseq assembly support. A subset of 39,050 predicted proteins have EST support over 80% of the mRNA, and13,568 gene models correspond to full lengthA. thaliana proteins.
Our sunflower genome database displays the reference genome sequence, annotation, and links to the genetic and physical maps. The primary entry point to the genome data is a genome browser (JBrowse), which displays the raw genome sequence along with various data tracks. These tracks include aligned and annotated transcripts, the scaffolds from which the assembly was derived, as well as SNP and expression data. The transcripts are cross-linked to the closest similar gene in external databases. The scaffolds are cross-linked to their position on the genetic map. The genetic map data are displayed in cMap, which in turn are linked to the corresponding physical genome region in JBrowse. The genome browser also supports searching based on physical regions, or known names of scaffolds or transcripts. We also offer a local BLAST search against the genome assembly, with search hits being linked to the corresponding genome regions in the genome browser.
The second set of gene predictions we incorporated were derived from AUGUSTUS. Evidence-based hints for AUGUSTUS were produced with Scipio, which uses BLAT internally to align proteins (in our case, all non-hypothetical proteins in plant RefSeq) to the genome to infer splice sites.
The third step in our pipeline (depicted below) involves running MAKER-P with a set of proteins (again, plant RefSeq) and an assembled transcriptome to generate an initial set of gene predictions based on these alignments alone. These initial predictions from MAKER-P were used to train the gene prediction program SNAP.
Assimilating predictions - production of gene models
The fourth step is to run MAKER-P again with the following evidence:
- Transcriptome and protein alignments from step three above
- Gene predictions from EuGene
- Gene predictions from AUGUSTUS
- Gene predictions from SNAP
An important difference in step four is that we tell MAKER to not build gene models from alignment evidence alone. With the evidence from step four we re-train SNAP.
The final prediction step in our annotation pipeline is aimed at incorporating lowly-expressed and alternatively spliced genes. To that end, we generated assembled transcripts with StringTie from 10 tissue-specific transcriptome libraries.
Final step - re-training models
In the final modeling phase we incorporated the following evidence:
- Transcriptome and protein alignments from step three above
- Gene predictions from EuGene
- Gene predictions from AUGUSTUS
- Gene predictions from the second round of training SNAP
- Transcript assemblies from StringTie
The gene models are available in a variety of formats (e.g., GFF3, GTF, FASTA) along with a plain text file with descriptive statistics about the models in the downloads section. On that page you can also find functional annotations of the genes, which are incorporated as attributes of the features in the GFF3 and GTF files.